
|
|
|
|
||
(Circulation. 2002;106:2760.)
© 2002 American Heart Association, Inc.
Editorial |
Peter Libby, MD; Jorge Plutzky, MD
From The Leducq Center for Cardiovascular Research, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Mass.
Correspondence to Peter Libby, MD, The Leducq Center for Cardiovascular Research, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, 221 Longwood Ave, EBRC 307, Boston, MA 02115. E-mail plibby@rics.bwh.harvard.edu
Key Words: Editorials • atherosclerosis
• diabetes mellitus • lipid • risk factors
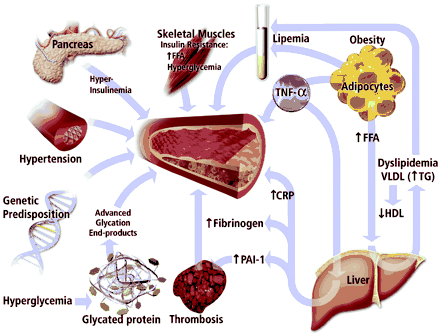
The cardiology community is awakening to a rampant epidemic of type II diabetes and its common companion, the metabolic syndrome. As the ponderosity of the US population increases, the morbid constellation of obesity, hypertension, glucose intolerance, insulin resistance, and dyslipidemia (characterized by abundant triglyceride (TG)–rich lipoproteins, low levels of atheroprotective high-density lipoprotein [HDL], and small, dense low-density lipoprotein [LDL] particles) is on the rise.1 Pioneering work from several laboratories has provided us with pathophysiological insight for understanding some of diabetes’ vascular complications. In the face of hyperglycemia, glucose molecules conjugate by a nonenzymatic mechanism with the reactive side chains of the amino acid lysine on protein molecules (Figure). Through a series of well-understood chemical reactions, this nonenzymatic glycation can ultimately generate higher molecular weight condensates known as advanced glycation end products (AGE).2,3 The formation of caramel from sugar provides a simple analogy for this process. Such reactions can be quite pervasive—occurring both inside and outside the cell, chemically modifying and potentially altering the functions not just of proteins, but of lipids and nucleic acids as well.
|
| Multiple
mechanisms contribute to arterial disease in patients
with type II diabetes. A variety of risk factors converge
on the artery to promote atherogenesis in individuals
with type II diabetes (center). Skeletal muscles may be
resistant of insulin action, which decreases the
utilization of glucose and free fatty acids, causing
hyperglycemia and increased levels of circulating free
fatty acids. In the face of the insulin resistance, the
pancreas initially attempts to compensate by producing
more insulin, yielding hyperinsulinemia, itself a risk
factor for arteriopathy. A high burden of abdominal fat
presents the liver with elevated levels of free fatty
acids through the portal circulation. This excess of free
fatty acids will drive the overproduction of TG-rich
lipoprotein particles, including VLDL. A reciprocal
decrease in HDL accompanies the hypertriglyceridemia
characteristic of the type II diabetic state. In addition
to the increase in fasting TGs, patients with diabetes
may have an accentuated response to dietary fat, yielding
an exaggerated postprandial lipemia, indicated by the
creamy supernatant over the plasma in the test tube. The
adipocyte can also release proinflammatory cytokines such
as TNF- |
See p 2827
Researchers have recognized the buildup of AGE-modified macromolecules for many years. However, recent discoveries have furnished a novel link between AGE-modified proteins and altered behavior of cells involved in arterial disease. Stern and colleagues characterized a cell surface receptor for AGE (RAGE).2 A number of groups have shown that engagement of RAGE can activate inflammatory functions of endothelial cells, smooth muscle cells, and macrophages, cell types intimately involved in atherogenesis. Engagement of RAGE can also increase oxidative stress. In addition to binding AGE, RAGE can bind cytokines of the S100/calgranulin family, providing another link between RAGE expression and inflammation, a process we now recognize as fundamental in the creation and complication of atherosclerotic lesions.
Previously, Schmidt, Stern, and colleagues demonstrated that interrupting AGE signaling in atherosclerosis-prone mice by infusing a decoy, a soluble form RAGE, decreased the formation of new atheroma. In the present issue of Circulation, this group now shows that administration of soluble RAGE can arrest the progression of already established atheroma.4 This treatment reduces the size of lesions and also changes qualitative characteristics of plaques that indicate reduced inflammation and increased "stability."5 Inhibition of RAGE signaling decreased levels of matrix-degrading proteinases and increased levels of interstitial collagen, the crucial protector of the integrity of the plaque’s fibrous cap. These important new experiments not only advance our knowledge of the pathophysiology of experimental atherosclerosis in these diabetic, atherosclerosis-prone mice, but also point to a new therapeutic target of considerable interest, given the epidemic of diabetic vascular disease we now confront.
Formation of AGE presumably relates to the level of glycemia. Indeed, our commonly used clinical index of glycemic control, hemoglobin A1C, measures a protein (hemoglobin) that has undergone nonenzymatic glycation, and correlates with AGE levels. Treatments that lower blood sugar reduce the level of this indicator-glycated protein. Given this link between glycemic control and ligands for RAGE, one might logically assume that strict glycemic control would protect against diabetic vascular complications. Indeed, several important clinical trials have demonstrated that stringent glycemic control significantly reduces the incidence of microvascular complications of diabetes such as nephropathy, retinopathy, and neuropathy.6–9
However, the plausible hypothesis that tight glycemic control would likewise reduce the risk of macrovascular complications of diabetes such as myocardial infarction has thus far eluded broad clinical proof. A number of well-conducted clinical trials, such as the University Group Diabetes Program (UGDP) and the United Kingdom Prospective Diabetes Study (UKPDS), among others, have found only limited, if any, relationship between glycemic control and diabetic macrovascular manifestations (Table 1).6–9 In stark contrast, numerous studies consistently show that pharmacological interventions that target the dyslipidemia and hypertension associated with type II diabetes can handily reduce risk of macrovascular complications in such patients. Thus, the goal of proving that glycemic control can also lower risk of heart attack or stroke still seems out of reach.8,9
TABLE 1. Cardiovascular Risk
Reduction in Patients With Diabetes According to Targeted Risk
Factor
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 2. Some Potential Contributors to the Glucose Paradox
|
Clinical trials indicate that strict glycemic control forestalls microvascular disease to a greater extent than macrovascular manifestations. Multiple factors may contribute to this disparity (Table 2). The studies conducted so far may well have just lacked sufficient power to settle the question, as they often show a trend for decreased cardiovascular events but fall short of achieving statistical significance. Indeed, the intensive antidiabetic treatment arm in the UKPDS reported a 16% reduction in myocardial infarction (MI) (P=0.052). Even if underpowering contributes to this possible glucose paradox, it appears that current antidiabetic treatments do not match the impact of treatments like statins or interruption of angiotensin II signaling (Table 1).
The specific interventions used to lower glycemia may also
contribute to the inability to show decreases in
macrovascular end points. With some antidiabetic
treatments, untoward effects may counterbalance potential
benefits. Generally, interventions that increase insulin supply
(eg, insulin itself and sulfonylureas) have proven less promising
for limiting cardiovascular complications than those that
improve glucose utilization or reduce insulin resistance. Indeed,
in one arm of UKPDS, metformin monotherapy decreased MI
by 39% (P![]() 0.01) in an overweight subgroup, a benefit
not seen in patients requiring metformin plus
sulfonylureas or insulin.10
Thiazolidinediones (the "glitazones") hold
considerable promise as insulin sensitizers and merit
careful clinical evaluation for cardiovascular
benefit.11
0.01) in an overweight subgroup, a benefit
not seen in patients requiring metformin plus
sulfonylureas or insulin.10
Thiazolidinediones (the "glitazones") hold
considerable promise as insulin sensitizers and merit
careful clinical evaluation for cardiovascular
benefit.11
Perhaps too short a duration or too tardy institution of better glycemic control accounts for the lack of effect on end points related to atherosclerosis in patients with diabetes. We know that the metabolic derangements in type II diabetes precede the development of frank diabetes by many years. Thus, hyperglycemia may have gradually wrought its damage over time in such a way that the duration of intervention afforded in clinical trials does not suffice to reverse its ravages. However, over a similar duration of treatment (3 to 5 years), other interventions can reduce macrovascular events, as shown with statins, fibrates, and agents that disrupt angiotensin II signaling.
The expectation that strict glycemic control alone can
mitigate atherosclerosis in type II diabetes does not
take into account the multiplicity of contributory
metabolic and inflammatory factors (Figure).
Adipose tissue itself can release proinflammatory stimuli
that may well produce "echoes" at the level of the
artery wall.12
Moreover, tumor necrosis factor-![]() and other proinflammatory cytokines
produced by adipocytes can increase production in the
liver of fibrinogen and plasminogen activator inhibitor, tipping
the hemostatic balance in the vessel wall toward thrombosis.
These cytokines can beget the production of C-reactive
protein from hepatocytes. C-reactive protein may not
only mark the risk of vascular complications, but may
also participate as a proinflammatory mediator13
and even predict onset of new diabetes.14
and other proinflammatory cytokines
produced by adipocytes can increase production in the
liver of fibrinogen and plasminogen activator inhibitor, tipping
the hemostatic balance in the vessel wall toward thrombosis.
These cytokines can beget the production of C-reactive
protein from hepatocytes. C-reactive protein may not
only mark the risk of vascular complications, but may
also participate as a proinflammatory mediator13
and even predict onset of new diabetes.14
Moreover, the complex pattern of dyslipidemia commonly
encountered in type II diabetes may also promote
arterial inflammation and hence atherogenesis.
Although patients with type II diabetes often have
average levels of LDL, they typically have qualitative abnormalities
in these particles. The small, dense LDL typical of
type II diabetes has particular susceptibility to oxidative modification
and, therefore, triggering of inflammation. The TG-rich
lipoproteins, such as ß-very-low-density lipoprotein, may
also incite inflammation by activating the transcription factor
NF-![]() B, an orchestrator of the
expression of proinflammatory genes related to
atherogenesis.15
Low levels of HDL rob the vessel wall of a protective
particle that promotes efflux of lipid from the
arterial wall and carries antioxidant enzymes. Thus,
the multifactorial complexity of diabetic vascular disease may
stymie the ability of strict glycemic control to forestall atherosclerotic
events. Although the study of Bucciarelli et al4
suggests an important role for RAGE in progression of atheroma,
we must nonetheless acknowledge that the management of
diabetic macrovascular disease requires much more than
attention to glycemia.
B, an orchestrator of the
expression of proinflammatory genes related to
atherogenesis.15
Low levels of HDL rob the vessel wall of a protective
particle that promotes efflux of lipid from the
arterial wall and carries antioxidant enzymes. Thus,
the multifactorial complexity of diabetic vascular disease may
stymie the ability of strict glycemic control to forestall atherosclerotic
events. Although the study of Bucciarelli et al4
suggests an important role for RAGE in progression of atheroma,
we must nonetheless acknowledge that the management of
diabetic macrovascular disease requires much more than
attention to glycemia.
Although we look forward to ongoing and future trials with existing antidiabetic drugs and the development of new treatments for diabetic macrovascular disease, we must not forget to implement therapies known today to prevent vascular complications of diabetes. Proven strategies include addressing the prothrombotic state with aspirin, treating dyslipidemia to values targeted by national guidelines, and achieving blood pressure goals of 130/85 mm Hg as mandated by the American Diabetes Association.9 Nonpharmacological lifestyle modifications, although hard to achieve in practice, can impressively improve metabolic variables in type II diabetes correlated with cardiovascular events. On the basis of exciting and novel research avenues such as those represented by the work of Bucciarelli et al,4 we may look forward to an "age of AGE" as a future target of therapy. In addition to a glucose paradox, we confront a "treatment paradox": insufficient adoption of therapies that can improve macrovascular end points in diabetes. Although we await tomorrow’s advances, we must implement today our current preventive guidelines with intensified fervor to reduce the growing burden of cardiovascular morbidity and mortality among patients with diabetes.
Footnotes
The opinions expressed in this editorial are not necessarily those of the editors or of the American Heart Association.
References
 35%
35%