|
|
|
|
|
|
Reversal of Obesity- and Diet-Induced Insulin Resistance with Salicylates
or Targeted Disruption of Ikk
Minsheng Yuan,1*
Nicky Konstantopoulos,1* Jongsoon Lee,1* Lone Hansen,1
Zhi-Wei Li,2 Michael Karin,2 Steven E. Shoelson1![]()
We show that high doses of
salicylates reverse hyperglycemia, hyperinsulinemia, and dyslipidemia in obese
rodents by sensitizing insulin signaling. Activation or
overexpression of the I![]() B
kinase
B
kinase ![]() (IKK
(IKK![]() )
attenuated insulin signaling in cultured cells, whereas IKK
)
attenuated insulin signaling in cultured cells, whereas IKK![]() inhibition reversed insulin resistance. Thus, IKK
inhibition reversed insulin resistance. Thus, IKK![]() ,
rather than the cyclooxygenases, appears to be the relevant
molecular target. Heterozygous deletion (Ikk
,
rather than the cyclooxygenases, appears to be the relevant
molecular target. Heterozygous deletion (Ikk![]() +/
+/![]() )
protected against the development of insulin resistance during high-fat
feeding and in obese Lepob/ob mice. These findings implicate
an inflammatory process in the pathogenesis of insulin resistance in
obesity and type 2 diabetes mellitus and identify the IKK
)
protected against the development of insulin resistance during high-fat
feeding and in obese Lepob/ob mice. These findings implicate
an inflammatory process in the pathogenesis of insulin resistance in
obesity and type 2 diabetes mellitus and identify the IKK![]() pathway as a target for insulin sensitization.
pathway as a target for insulin sensitization.
1 Joslin Diabetes Center and Department of Medicine, Harvard
Medical School, Boston, MA 02215, USA.
2 Department of Pharmacology, University of California, San Diego,
La Jolla, CA 92093, USA.
* These authors contributed equally to this work.
![]() To whom correspondence should be addressed. E-mail: Steven.Shoelson@Joslin.Harvard.edu
To whom correspondence should be addressed. E-mail: Steven.Shoelson@Joslin.Harvard.edu
Insulin resistance refers to a decreased capacity of
circulating insulin to regulate nutrient metabolism. Individuals with insulin
resistance are predisposed to developing type 2 diabetes, and
insulin resistance is an integral feature of its pathophysiology. Chronic
secretion of large amounts of insulin to overcome tissue insensitivity
can lead, in predisposed individuals, to pancreatic ![]() cell
failure and concomitant defects in glucose and lipid metabolism. The
prevalence of insulin resistance is high and rising, but only rare
genetic causes have been identified. The molecular cause of acquired
insulin resistance, which is promoted by sedentary lifestyle,
obesity, fatty diet, and increased age, and is reversed by exercise
and weight loss, is similarly unknown.
cell
failure and concomitant defects in glucose and lipid metabolism. The
prevalence of insulin resistance is high and rising, but only rare
genetic causes have been identified. The molecular cause of acquired
insulin resistance, which is promoted by sedentary lifestyle,
obesity, fatty diet, and increased age, and is reversed by exercise
and weight loss, is similarly unknown.
High doses of salicylates [4 to
10 g per day (g/day)], including sodium salicylate and aspirin, have been
used to treat inflammatory conditions such as rheumatic fever and
rheumatoid arthritis. These high doses are thought to inhibit
nuclear factor kappa B (NF-![]() B)
(1)
and its upstream activator the I
B)
(1)
and its upstream activator the I![]() B
kinase
B
kinase ![]() (IKK
(IKK![]() )
(2),
as opposed to working through cyclooxygenases (COXs), the classical
targets of nonsteroidal anti-inflammatory drugs (NSAIDs). High doses
of salicylates also lower blood glucose concentrations (3-7),
although their potential for treating diabetes has been all but
forgotten by modern biomedical science. We have investigated
potential mechanisms of these hypoglycemic effects to identify
potential mediators of insulin resistance and molecular targets for
intervention. We have found that reduced signaling through the IKK
)
(2),
as opposed to working through cyclooxygenases (COXs), the classical
targets of nonsteroidal anti-inflammatory drugs (NSAIDs). High doses
of salicylates also lower blood glucose concentrations (3-7),
although their potential for treating diabetes has been all but
forgotten by modern biomedical science. We have investigated
potential mechanisms of these hypoglycemic effects to identify
potential mediators of insulin resistance and molecular targets for
intervention. We have found that reduced signaling through the IKK![]() pathway, either by salicylate inhibition or decreased IKK
pathway, either by salicylate inhibition or decreased IKK![]() expression, is accompanied by improved insulin sensitivity in vivo.
Our findings further indicate that the IKK
expression, is accompanied by improved insulin sensitivity in vivo.
Our findings further indicate that the IKK![]() pathway may contribute to insulin resistance in type 2 diabetes
and obesity by impinging on insulin signaling.
pathway may contribute to insulin resistance in type 2 diabetes
and obesity by impinging on insulin signaling.
We determined the effect of high doses of salicylates on the severe insulin resistance seen in genetically obese rodents. Twelve-week-old male Zucker fa/fa rats and 8-week-old male ob/ob mice were treated for 3 to 4 weeks with 120 mg/kg/day of aspirin or sodium salicylate, administered by continuous subcutaneous infusion. Fasting blood glucose values and glucose tolerance were improved in Zucker fa/fa rats (Fig. 1A). Concomitant reductions in insulin concentrations (Fig. 1B) indicated a marked improvement in insulin sensitivity. Glucose tolerance in lean fa/+ animals was normal, and blood glucose concentrations were similar after aspirin treatment (Fig. 1C). Nevertheless, lower insulin concentrations in the aspirin-treated group (Fig. 1D) demonstrate improved insulin sensitivity, despite milder insulin resistance. The ability of high-dose aspirin to increase insulin sensitivity was further established in insulin tolerance tests (Fig. 1E). Intraperitoneal injection of insulin (2.0 U per kilogram of body weight) had essentially no effect on blood glucose concentrations in untreated fa/fa rats. However, the same insulin dose caused a decrease in blood glucose when given to aspirin-treated animals.
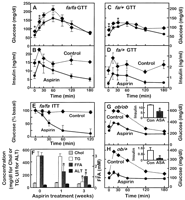 Fig.
1. In vivo effects of aspirin in Zucker fatty rats and ob/ob mice.
(A to F) Twelve-week-old male Zucker fa/fa rats and (G and
H) 8-week-old ob/ob (Lepob/ob) and ob/+
mice were given free access to food and water. Aspirin (120 mg/kg/day) was
continuously infused for 3 to 4 weeks by Alzet pumps (pump 2ML2 was
used in rats, pump 2002 was used in mice) implanted subcutaneously between
the scapulae of the animals. In all panels, data are mean ± SEM values,
diamonds and dashed lines represent control implantation of a pump with vehicle
only, and solid circles and solid lines represent 3 or 4 weeks of
treatment with aspirin. For glucose tolerance tests (GTT), glucose
(2.0 g/kg) was administered by oral gavage (rats) or intraperitoneal
injection (mice) after an overnight fast. (A and C) Blood glucose and (B and D)
serum insulin concentrations were determined during oral glucose tolerance
tests in (A and B) Zucker fa/fa rats or (C and D) fa/+ rats; six
animals were in each treatment group. In (A) and (C), *P < 0.05, **P
< 0.01 for vehicle versus aspirin treatment, Student's t test.
(B) P = 0.0004 and (D) P = 0.02, for
vehicle versus aspirin treatment, integrated areas under the curves; Student's
t test. (E) For insulin tolerance tests (ITT), insulin (2.0 U/kg) was
injected intraperitoneally after an overnight fast (six Zucker fa/fa
rats, P = 0.0001, for vehicle versus aspirin treatment,
integrated areas under the curves; Student's t test). (F) Cholesterol
(Chol), triglyceride (TG), long-chain FFA, and ALT concentrations were measured
in sera from fasting Zucker fa/fa rats (for FFA, P < 0.005 for
three-group analysis of variance; *P = 0.01, t = 0 versus
t = 1 week of aspirin treatment, **P < 0.002, t = 0 versus
t = 3 weeks of aspirin treatment, Dunnett's adjusted
pairwise comparison. For TG, *P <0.05, t = 0 versus
t = 3 weeks of aspirin treatment; Student's t tests).
Glucose tolerance tests are shown in (G) ob/ob and (H) ob/+ mice.
(G) P = 0.004 and (H) P < 0.0001 for
10 mice treated with vehicle versus 10 mice treated with aspirin,
integrated areas under the curves; Student's t tests. [Insets in (G) and
(H)] Fasting serum insulin concentrations (ng/ml); **P < 0.01 for
10 mice treated with aspirin versus 10 controls treated with vehicle
alone. Aspirin treatment did not significantly affect food intake or body
weights in any of these studies. Animal care was in accordance with
institutional and NIH guidelines. [View Larger
Version of this Image (57K GIF file)]
Fig.
1. In vivo effects of aspirin in Zucker fatty rats and ob/ob mice.
(A to F) Twelve-week-old male Zucker fa/fa rats and (G and
H) 8-week-old ob/ob (Lepob/ob) and ob/+
mice were given free access to food and water. Aspirin (120 mg/kg/day) was
continuously infused for 3 to 4 weeks by Alzet pumps (pump 2ML2 was
used in rats, pump 2002 was used in mice) implanted subcutaneously between
the scapulae of the animals. In all panels, data are mean ± SEM values,
diamonds and dashed lines represent control implantation of a pump with vehicle
only, and solid circles and solid lines represent 3 or 4 weeks of
treatment with aspirin. For glucose tolerance tests (GTT), glucose
(2.0 g/kg) was administered by oral gavage (rats) or intraperitoneal
injection (mice) after an overnight fast. (A and C) Blood glucose and (B and D)
serum insulin concentrations were determined during oral glucose tolerance
tests in (A and B) Zucker fa/fa rats or (C and D) fa/+ rats; six
animals were in each treatment group. In (A) and (C), *P < 0.05, **P
< 0.01 for vehicle versus aspirin treatment, Student's t test.
(B) P = 0.0004 and (D) P = 0.02, for
vehicle versus aspirin treatment, integrated areas under the curves; Student's
t test. (E) For insulin tolerance tests (ITT), insulin (2.0 U/kg) was
injected intraperitoneally after an overnight fast (six Zucker fa/fa
rats, P = 0.0001, for vehicle versus aspirin treatment,
integrated areas under the curves; Student's t test). (F) Cholesterol
(Chol), triglyceride (TG), long-chain FFA, and ALT concentrations were measured
in sera from fasting Zucker fa/fa rats (for FFA, P < 0.005 for
three-group analysis of variance; *P = 0.01, t = 0 versus
t = 1 week of aspirin treatment, **P < 0.002, t = 0 versus
t = 3 weeks of aspirin treatment, Dunnett's adjusted
pairwise comparison. For TG, *P <0.05, t = 0 versus
t = 3 weeks of aspirin treatment; Student's t tests).
Glucose tolerance tests are shown in (G) ob/ob and (H) ob/+ mice.
(G) P = 0.004 and (H) P < 0.0001 for
10 mice treated with vehicle versus 10 mice treated with aspirin,
integrated areas under the curves; Student's t tests. [Insets in (G) and
(H)] Fasting serum insulin concentrations (ng/ml); **P < 0.01 for
10 mice treated with aspirin versus 10 controls treated with vehicle
alone. Aspirin treatment did not significantly affect food intake or body
weights in any of these studies. Animal care was in accordance with
institutional and NIH guidelines. [View Larger
Version of this Image (57K GIF file)]
Increased triglyceride concentrations in the blood of Zucker rats fell from 494 ± 68 mg/dl to 90 ± 58 mg/dl during 3 weeks of aspirin treatment (Fig. 1F). The concentrations of free fatty acid (FFA) dropped as well, from 3.1 ± 0.3 mM to 1.1 ± 0.2 mM. The decrease in the amount of circulating FFA occurred within 1 week of aspirin treatment, preceding reductions in the amounts of triglyceride and glucose in the blood. This is consistent with the hypothesis that increased FFA concentrations contribute to the pathogenesis of hyperglycemia and hypertriglyceridemia. Cholesterol concentrations were unaffected. Hepatotoxicity was not observed at the high aspirin and salicylate doses used, judging from the consistently normal circulating concentrations of the liver enzyme alanine aminotransferase (ALT), seen throughout our studies (Fig. 1F). Serum salicylate concentrations were 0.81 ± 0.25 mM.
Ob/ob mice (Lepob/ob) are a more relevant model for type 2 diabetes, as the animals are diabetic in addition to being obese and severely insulin-resistant. Fasting blood glucose values and glucose tolerance were significantly improved by aspirin treatment (Fig. 1G). Heterozygous, lean Lepob/+ mice exhibited postprandial hyperglycemia but were less insulin resistant. Glucose intolerance in Lepob/+ mice was normalized with aspirin treatment (Fig. 1H). Insulin concentrations were reduced during aspirin therapy in both Lepob/ob and Lepob/+ mice. Neither aspirin nor salicylate affected food intake or body weight in Zucker fa/fa rats or ob/ob mice.
We isolated tissues from treated animals to analyze various signaling proteins (8-10). Insulin receptor (IR) tyrosine phosphorylation, one of the earliest responses to insulin binding, was barely detectable in the liver and muscle of insulin-resistant Zucker rats (Fig. 2, A and B). Increased stimulation occurred in corresponding tissues from aspirin- and salicylate-treated animals, suggesting an increase in insulin responsiveness. Signaling from the IR to insulin receptor substrates (IRSs), phosphatidylinositol 3-kinase, and the PDK1 protein kinase is required for the maintenance of metabolic homeostasis. Phosphorylation of the protein kinase AKT, a subsequent step in this cascade, correlates with IR activation in tissues from Zucker rats (Fig. 2, A and B). The blunted insulin-stimulated phosphorylation of AKT in the liver and muscle of untreated Zucker rats was increased after aspirin or salicylate treatment, providing a biochemical correlate for increased in vivo insulin sensitivity. The electrophoretic mobility of IRS-1 from rat livers increased with aspirin and salicylate treatment [Web fig. 1 (11)], suggesting a decrease in serine-threonine (Ser-Thr) phosphorylation (this is a known inhibitor of insulin signaling). Basal IKK activity was elevated in tissues from Zucker fa/fa rats relative to those from lean fa/+ controls (12).
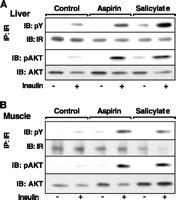 Fig.
2. Signaling proteins in tissues from aspirin- and salicylate-treated
Zucker fa/fa fatty rats. Animals treated for 3 to 4 weeks with
aspirin or sodium salicylate received intravenous injections of insulin or
saline 7 min before being killed. Tissues were harvested and frozen
immediately in liquid nitrogen. Liver and plantaris muscle samples were
homogenized and signaling proteins were identified by Western blotting (13). (A)
Liver or (B) muscle homogenates were immunoprecipitated with antiIR and
proteins were identified by blotting with anti-pY and anti-IR. Alternatively,
tissue homogenates were separated by SDS-PAGE and proteins were identified by
blotting with antibodies specific to phospho-AKT (pAKT) and antibodies to AKT.
IB, immunoblot; IP, immunoprecipitate. [View Larger
Version of this Image (67K GIF file)]
Fig.
2. Signaling proteins in tissues from aspirin- and salicylate-treated
Zucker fa/fa fatty rats. Animals treated for 3 to 4 weeks with
aspirin or sodium salicylate received intravenous injections of insulin or
saline 7 min before being killed. Tissues were harvested and frozen
immediately in liquid nitrogen. Liver and plantaris muscle samples were
homogenized and signaling proteins were identified by Western blotting (13). (A)
Liver or (B) muscle homogenates were immunoprecipitated with antiIR and
proteins were identified by blotting with anti-pY and anti-IR. Alternatively,
tissue homogenates were separated by SDS-PAGE and proteins were identified by
blotting with antibodies specific to phospho-AKT (pAKT) and antibodies to AKT.
IB, immunoblot; IP, immunoprecipitate. [View Larger
Version of this Image (67K GIF file)]
Studies with cultured cells were
used to investigate potential mechanisms relating salicylate treatment to the
in vivo reversal of insulin resistance. Treatment of 3T3-L1
adipocytes with tumor necrosis factor-![]() (TNF-
(TNF-![]() )
induced "insulin resistance," as judged by decreases in
insulin-stimulated tyrosine phosphorylation of the IR
)
induced "insulin resistance," as judged by decreases in
insulin-stimulated tyrosine phosphorylation of the IR ![]() -subunit
and IRS-1, to 42 ± 11% and 37 ± 9%, respectively, of
that in insulin-stimulated cells not treated with TNF-
-subunit
and IRS-1, to 42 ± 11% and 37 ± 9%, respectively, of
that in insulin-stimulated cells not treated with TNF-![]() (Fig. 3,
A and B) (13). This
was reversed by prior treatment of cells with high doses of aspirin
(5 mM). The amounts of IR and IRS-1 proteins were similar in
all cells. TNF-
(Fig. 3,
A and B) (13). This
was reversed by prior treatment of cells with high doses of aspirin
(5 mM). The amounts of IR and IRS-1 proteins were similar in
all cells. TNF-![]() activates the IKK complex (14).
Phosphatase inhibitors such as okadaic acid and calyculin A also
activate IKK
activates the IKK complex (14).
Phosphatase inhibitors such as okadaic acid and calyculin A also
activate IKK![]() (15,
16),
but without activating upstream elements in the TNF-
(15,
16),
but without activating upstream elements in the TNF-![]() signaling cascade, and these inhibitors also induce insulin resistance
in isolated tissues and cultured cells (17, 18).
Calyculin A reduced insulin-stimulated tyrosine phosphorylation of
the IR and IRS-1, to 29 ± 12% and 16 ± 2%, respectively,
of that in untreated cells, and this was prevented by incubating the
cells with aspirin (Fig. 3, A
and B). Similar results were obtained after okadaic acid treatment (19). The
reduced electrophoretic mobility of IRS-1 due to calyculin A
treatment was reversed with aspirin (Fig. 3C),
further suggesting that aspirin's ability to reverse insulin
resistance might result from reduced levels of Ser-Thr
phosphorylation of components in the insulin action cascade.
signaling cascade, and these inhibitors also induce insulin resistance
in isolated tissues and cultured cells (17, 18).
Calyculin A reduced insulin-stimulated tyrosine phosphorylation of
the IR and IRS-1, to 29 ± 12% and 16 ± 2%, respectively,
of that in untreated cells, and this was prevented by incubating the
cells with aspirin (Fig. 3, A
and B). Similar results were obtained after okadaic acid treatment (19). The
reduced electrophoretic mobility of IRS-1 due to calyculin A
treatment was reversed with aspirin (Fig. 3C),
further suggesting that aspirin's ability to reverse insulin
resistance might result from reduced levels of Ser-Thr
phosphorylation of components in the insulin action cascade.
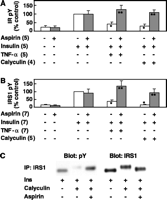 Fig.
3. Aspirin effects on insulin signaling in 3T3-L1 adipocytes. 3T3-L1
adipocytes were serum-starved for 16 hours and treated or not treated with
5 mM aspirin for 2 hours and either 6.0 nM mTNF-
Fig.
3. Aspirin effects on insulin signaling in 3T3-L1 adipocytes. 3T3-L1
adipocytes were serum-starved for 16 hours and treated or not treated with
5 mM aspirin for 2 hours and either 6.0 nM mTNF-![]() (20 min) or the phosphatase inhibitor calyculin A (at 2.0 nM for
30 min). After a 5-min stimulation with 10 nM insulin, the cells were
chilled and solubilized and proteins were immunoprecipitated with (A)
anti-IR or (B and C) anti-IRS1. Proteins separated by SDS-PAGE
were identified by Western blotting with anti-pY, anti-IR, or anti-IRS1.
Control blots (not shown) indicated that protein amounts of IR and IRS1 did not
differ significantly between treatments. Ins, insulin. (A and B)
Phosphorylation levels were quantified by densitometry and expressed relative
to insulin-stimulated controls; the numbers of individual experiments are shown
in parentheses (*P < 0.01 for minus aspirin, plus
insulin, minus versus plus mTNF-
(20 min) or the phosphatase inhibitor calyculin A (at 2.0 nM for
30 min). After a 5-min stimulation with 10 nM insulin, the cells were
chilled and solubilized and proteins were immunoprecipitated with (A)
anti-IR or (B and C) anti-IRS1. Proteins separated by SDS-PAGE
were identified by Western blotting with anti-pY, anti-IR, or anti-IRS1.
Control blots (not shown) indicated that protein amounts of IR and IRS1 did not
differ significantly between treatments. Ins, insulin. (A and B)
Phosphorylation levels were quantified by densitometry and expressed relative
to insulin-stimulated controls; the numbers of individual experiments are shown
in parentheses (*P < 0.01 for minus aspirin, plus
insulin, minus versus plus mTNF-![]() ,
or minus versus plus calyculin A; **P < 0.05 for minus
versus plus aspirin of mTNF-
,
or minus versus plus calyculin A; **P < 0.05 for minus
versus plus aspirin of mTNF-![]() or calyculin A-treated pairs; Student's t test). [View Larger
Version of this Image (27K GIF file)]
or calyculin A-treated pairs; Student's t test). [View Larger
Version of this Image (27K GIF file)]
Fao hepatoma cells are an
insulin-responsive model for liver as opposed to fat. TNF-![]() treatment decreased tyrosine phosphorylation of the insulin receptor
substrate IRS-2 (Fig. 4A) (20). Decreased
IRS-2 tyrosine phosphorylation was reversed by aspirin or sodium
salicylate. Aspirin and sodium salicylate are equipotent inhibitors
of IKK
treatment decreased tyrosine phosphorylation of the insulin receptor
substrate IRS-2 (Fig. 4A) (20). Decreased
IRS-2 tyrosine phosphorylation was reversed by aspirin or sodium
salicylate. Aspirin and sodium salicylate are equipotent inhibitors
of IKK![]() (2),
whereas aspirin is ~100-fold more potent toward the cyclooxygenases
(21).
Our findings suggest that IKK
(2),
whereas aspirin is ~100-fold more potent toward the cyclooxygenases
(21).
Our findings suggest that IKK![]() ,
and not COX1 nor COX2, might alter insulin signaling. Additional
NSAIDs were used to further evaluate potential molecular mediators.
Ibuprofen and naproxen, which inhibit both COX1 and COX2, did not
reverse TNF-
,
and not COX1 nor COX2, might alter insulin signaling. Additional
NSAIDs were used to further evaluate potential molecular mediators.
Ibuprofen and naproxen, which inhibit both COX1 and COX2, did not
reverse TNF-![]() -induced
insulin resistance (Fig. 4C).
The selective COX2 inhibitor NS-398 similarly had no effect. Studies
were conducted with doses of the drugs known to have biological
effects (2,
21).
These pharmacological profiles further point to IKK as the target of
these effects and demonstrate that COX1 and COX2, the classical targets
for NSAIDs, do not mediate the antidiabetic effects of aspirin and
salicylate. This issue was addressed further using mice with reduced
COX expression; homozygous and heterozygous deletions of either COX1
or COX2 had no effect on carbohydrate or lipid metabolism in
insulin-resistant mice (22).
-induced
insulin resistance (Fig. 4C).
The selective COX2 inhibitor NS-398 similarly had no effect. Studies
were conducted with doses of the drugs known to have biological
effects (2,
21).
These pharmacological profiles further point to IKK as the target of
these effects and demonstrate that COX1 and COX2, the classical targets
for NSAIDs, do not mediate the antidiabetic effects of aspirin and
salicylate. This issue was addressed further using mice with reduced
COX expression; homozygous and heterozygous deletions of either COX1
or COX2 had no effect on carbohydrate or lipid metabolism in
insulin-resistant mice (22).
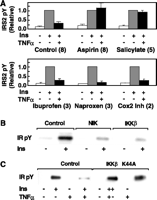 Fig.
4. (A) NSAID effects in Fao hepatoma cells. Fao cells were
serum-starved for 16 hours followed by 2-hour incubations at 37°C with
5 mM aspirin, 10 mM sodium salicylate, 25 µM ibuprofen,
25 µM sodium naproxen, or 25 µM NS-398 (a selective COX2 inhibitor).
Cells were then stimulated sequentially with 6.0 nM mTNF-
Fig.
4. (A) NSAID effects in Fao hepatoma cells. Fao cells were
serum-starved for 16 hours followed by 2-hour incubations at 37°C with
5 mM aspirin, 10 mM sodium salicylate, 25 µM ibuprofen,
25 µM sodium naproxen, or 25 µM NS-398 (a selective COX2 inhibitor).
Cells were then stimulated sequentially with 6.0 nM mTNF-![]() for 20 min and 10 nM insulin for 5 min. Cells were chilled and
solubilized and proteins were immunoprecipitated with anti-IR and detected by
Western blotting using anti-pY. Phosphorylation was quantified by densitometry
and is expressed relative to insulin-stimulated controls; the numbers of
individual experiments are shown in parentheses. (B and C)
Induction of insulin resistance with I
for 20 min and 10 nM insulin for 5 min. Cells were chilled and
solubilized and proteins were immunoprecipitated with anti-IR and detected by
Western blotting using anti-pY. Phosphorylation was quantified by densitometry
and is expressed relative to insulin-stimulated controls; the numbers of
individual experiments are shown in parentheses. (B and C)
Induction of insulin resistance with I![]() B
kinases and reversal with dominant inhibitors. (B) Myc-tagged NIK or
Flag-tagged IKK
B
kinases and reversal with dominant inhibitors. (B) Myc-tagged NIK or
Flag-tagged IKK![]() or (C) kinase-deficient IKK
or (C) kinase-deficient IKK![]() were expressed in HEK 293 cells. The pRK7 (cytomegalovirus) vectors
encoding the kinases were obtained from M. Rothe (Tularik); transfections
of 50 to 60% confluent cells (1.5 µg of DNA per well; six well
dishes) using FuGene 6 (Boehringer-Mannheim) were as recommended.
Experiments were initiated 36 hours after transfection and 16 hours
after removal of serum from the culture medium. (B) Cells were stimulated for
5 min with 10 nM insulin unless otherwise indicated. (C) Cells were
treated for 40 min with 6 nM mTNF-
were expressed in HEK 293 cells. The pRK7 (cytomegalovirus) vectors
encoding the kinases were obtained from M. Rothe (Tularik); transfections
of 50 to 60% confluent cells (1.5 µg of DNA per well; six well
dishes) using FuGene 6 (Boehringer-Mannheim) were as recommended.
Experiments were initiated 36 hours after transfection and 16 hours
after removal of serum from the culture medium. (B) Cells were stimulated for
5 min with 10 nM insulin unless otherwise indicated. (C) Cells were
treated for 40 min with 6 nM mTNF-![]() ,
followed by 5-min stimulations with 10 nM insulin. In (B) and (C), protein
expression levels ranged from 10 to 20 times that of the endogenous
proteins. [View
Larger Version of this Image (31K GIF file)]
,
followed by 5-min stimulations with 10 nM insulin. In (B) and (C), protein
expression levels ranged from 10 to 20 times that of the endogenous
proteins. [View
Larger Version of this Image (31K GIF file)]
IKK complexes contain the
heterodimeric kinases IKK![]() and IKK
and IKK![]() and the scaffolding protein IKK
and the scaffolding protein IKK![]() (14).
Either the IKK
(14).
Either the IKK![]() catalytic subunit or NIK, an upstream activator, was expressed in
HEK 293 cells. Insulin stimulated the activation of IR (Fig. 4B),
IRS-2, and AKT (19);
activation was attenuated by expression of IKK
catalytic subunit or NIK, an upstream activator, was expressed in
HEK 293 cells. Insulin stimulated the activation of IR (Fig. 4B),
IRS-2, and AKT (19);
activation was attenuated by expression of IKK![]() or NIK (Fig.
4B) [Web fig. 2 (11)], and
attenuated activation was reversed by treatment with aspirin (19). I
or NIK (Fig.
4B) [Web fig. 2 (11)], and
attenuated activation was reversed by treatment with aspirin (19). I![]() B
B![]() levels were reduced by IKK
levels were reduced by IKK![]() or NIK expression [Web fig. 2 (11)].
Because IKK
or NIK expression [Web fig. 2 (11)].
Because IKK![]() and IKK
and IKK![]() form heterodimers, overexpression of kinase-deficient IKK
form heterodimers, overexpression of kinase-deficient IKK![]() or IKK
or IKK![]() inhibits endogenous components of the complex and signaling to NF-
inhibits endogenous components of the complex and signaling to NF-![]() B
(23).
We therefore asked whether the expression of dominant inhibitory IKK
B
(23).
We therefore asked whether the expression of dominant inhibitory IKK![]() (K44A)
blocked the induction of insulin resistance. TNF-
(K44A)
blocked the induction of insulin resistance. TNF-![]() treatment reduced insulin-stimulated IR activation to 29 ± 2%
of untreated controls, and expression of IKK
treatment reduced insulin-stimulated IR activation to 29 ± 2%
of untreated controls, and expression of IKK![]() (K44A)
reversed the TNF-
(K44A)
reversed the TNF-![]() -inhibited
effects (Fig.
4C) [Web fig. 2 (11)]. Active
IKK
-inhibited
effects (Fig.
4C) [Web fig. 2 (11)]. Active
IKK![]() thus promotes insulin resistance in cultured cells, and the
inactive, dominant inhibitory kinase blocks TNF-
thus promotes insulin resistance in cultured cells, and the
inactive, dominant inhibitory kinase blocks TNF-![]() induced insulin resistance.
induced insulin resistance.
Studies with mice having targeted
disruption of the Ikk![]() locus further tested potential roles of IKK
locus further tested potential roles of IKK![]() in the development and reversal of insulin resistance. Ikk
in the development and reversal of insulin resistance. Ikk![]()
![]() /
/![]() mice die in utero because of enhanced liver apoptosis, whereas heterozygous
Ikk
mice die in utero because of enhanced liver apoptosis, whereas heterozygous
Ikk![]() +/
+/![]() mice were reportedly normal (24, 25).
Fasting glucose and insulin concentrations were consistently lower
in Ikk
mice were reportedly normal (24, 25).
Fasting glucose and insulin concentrations were consistently lower
in Ikk![]() +/
+/![]() compared to Ikk
compared to Ikk![]() +/+
littermates (Fig.
5, A and B). The potential protective effect of reduced Ikk
+/+
littermates (Fig.
5, A and B). The potential protective effect of reduced Ikk![]() gene dose was tested in crosses between Ikk
gene dose was tested in crosses between Ikk![]() +/
+/![]() and Lepob/ob mice. Fasting blood glucose concentrations were
reduced in Ikk
and Lepob/ob mice. Fasting blood glucose concentrations were
reduced in Ikk![]() +/
+/![]() Lepob/ob
mice compared with those in Ikk
Lepob/ob
mice compared with those in Ikk![]() +/+Lepob/ob
littermates (Fig.
5C). Glucose tolerance in the Ikk
+/+Lepob/ob
littermates (Fig.
5C). Glucose tolerance in the Ikk![]() +/
+/![]() Lepob/ob
mice was improved compared to Ikk
Lepob/ob
mice was improved compared to Ikk![]() +/+Lepob/ob
littermates (Fig.
5D), although insulin concentrations were indistinguishable (12). These
findings are consistent with improved insulin sensitivity in Ikk
+/+Lepob/ob
littermates (Fig.
5D), although insulin concentrations were indistinguishable (12). These
findings are consistent with improved insulin sensitivity in Ikk![]() +/
+/![]() Lepob/ob
mice compared to that in Ikk
Lepob/ob
mice compared to that in Ikk![]() +/+Lepob/ob
littermates. Also consistent with improved metabolic control, plasma
FFA concentrations were lower in the Ikk
+/+Lepob/ob
littermates. Also consistent with improved metabolic control, plasma
FFA concentrations were lower in the Ikk![]() +/
+/![]() Lepob/ob
mice (1.86 ± 0.12 mM) than their Ikk
Lepob/ob
mice (1.86 ± 0.12 mM) than their Ikk![]() +/+Lepob/ob
littermates (2.24 ± 0.09 mM; P = 0.03).
+/+Lepob/ob
littermates (2.24 ± 0.09 mM; P = 0.03).
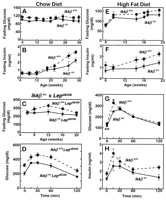 Fig.
5. Glucose tolerance and insulin sensitivity in Ikk
Fig.
5. Glucose tolerance and insulin sensitivity in Ikk![]() +/-
mice. (A and B) Ikk
+/-
mice. (A and B) Ikk![]() +/-
mice were backcrossed four generations on a C57BL/6J background. Fasting blood
glucose and fasting insulin concentrations were determined in Ikk
+/-
mice were backcrossed four generations on a C57BL/6J background. Fasting blood
glucose and fasting insulin concentrations were determined in Ikk![]() +/-
and Ikk
+/-
and Ikk![]() +/+
male littermates. Values represent the mean ± SEM for wild-type (WT)
(diamonds, n = 9) and Ikk+/- (circles, n = 6)
mice. (A) P <0.001 and (B) P = 0.02 for
integrated areas under the curves, Student's t tests for WT versus Ikk+/-
mice. (C and D) Ikk
+/+
male littermates. Values represent the mean ± SEM for wild-type (WT)
(diamonds, n = 9) and Ikk+/- (circles, n = 6)
mice. (A) P <0.001 and (B) P = 0.02 for
integrated areas under the curves, Student's t tests for WT versus Ikk+/-
mice. (C and D) Ikk![]() +/-
offspring were crossed with Lepob/+ (C57BL/6J) mice (Jackson
Laboratory, Bar Harbor, ME). F1 male Ikk+/- Lepob/+
offspring were crossed with Ikk+/+Lepob/+ females,
and F2 littermates were studied. (C) Fasting blood glucose
concentrations in male littermates. Values represent the mean ± SEM
for Ikk+/+Lepob/ob (diamonds, n = 7)
and Ikk+/-Lepob/ob (circles, n = 7);
P = 0.003 for integrated areas under the curves;
Student's t tests, Ikk+/-Lepob/ob versus
Ikk+/+ Lepob/ob. (D) Glucose tolerance tests were
conducted with 9- to 12-week-old male Ikk+/
+/-
offspring were crossed with Lepob/+ (C57BL/6J) mice (Jackson
Laboratory, Bar Harbor, ME). F1 male Ikk+/- Lepob/+
offspring were crossed with Ikk+/+Lepob/+ females,
and F2 littermates were studied. (C) Fasting blood glucose
concentrations in male littermates. Values represent the mean ± SEM
for Ikk+/+Lepob/ob (diamonds, n = 7)
and Ikk+/-Lepob/ob (circles, n = 7);
P = 0.003 for integrated areas under the curves;
Student's t tests, Ikk+/-Lepob/ob versus
Ikk+/+ Lepob/ob. (D) Glucose tolerance tests were
conducted with 9- to 12-week-old male Ikk+/![]() Lepob/ob
and Ikk+/+Lepob/ob littermates. Glucose
(2.0 g/kg) was administered by intraperitoneal injection after an
overnight fast. Values represent the mean ± SEM for Ikk+/+Lepob/ob
(diamonds, n = 12 males) and Ikk+/- Lepob/ob
(circles, n = 11 males; P = 0.006 for
integrated areas under the curves; Student's t tests, Ikk+/-Lepob/ob
versus Ikk+/+Lepob/ob). (E to H)
Beginning at 4 weeks of age, male Ikk
Lepob/ob
and Ikk+/+Lepob/ob littermates. Glucose
(2.0 g/kg) was administered by intraperitoneal injection after an
overnight fast. Values represent the mean ± SEM for Ikk+/+Lepob/ob
(diamonds, n = 12 males) and Ikk+/- Lepob/ob
(circles, n = 11 males; P = 0.006 for
integrated areas under the curves; Student's t tests, Ikk+/-Lepob/ob
versus Ikk+/+Lepob/ob). (E to H)
Beginning at 4 weeks of age, male Ikk![]() +/-
and Ikk
+/-
and Ikk![]() +/+
littermates were maintained on high-fat diets (Research Diets D12331; 58% of
calories from coconut oil). Fasting (E) glucose and (F) insulin concentrations
were measured from 8- to 23-week-old male littermates (diamonds, n = 7 Ikk+/+
and circles, n = 8 Ikk+/
+/+
littermates were maintained on high-fat diets (Research Diets D12331; 58% of
calories from coconut oil). Fasting (E) glucose and (F) insulin concentrations
were measured from 8- to 23-week-old male littermates (diamonds, n = 7 Ikk+/+
and circles, n = 8 Ikk+/![]() mice. (E) P <0.05 and (F) P = 0.01 for
integrated areas under the curves, Student's t tests for WT versus Ikk+/-
mice. (G and H) Glucose tolerance tests were conducted with 18-week-old male
littermates. Values represent the mean ± SEM for Ikk+/+
(diamonds, n = 10) and Ikk+/
mice. (E) P <0.05 and (F) P = 0.01 for
integrated areas under the curves, Student's t tests for WT versus Ikk+/-
mice. (G and H) Glucose tolerance tests were conducted with 18-week-old male
littermates. Values represent the mean ± SEM for Ikk+/+
(diamonds, n = 10) and Ikk+/![]() (circles, n = 10). (G) *P <0.005; **P
<0.01, Student's t tests for WT versus Ikk+/- mice.
(H) P = 0.03 for integrated areas under the curves,
Student's t tests for WT versus Ikk+/
(circles, n = 10). (G) *P <0.005; **P
<0.01, Student's t tests for WT versus Ikk+/- mice.
(H) P = 0.03 for integrated areas under the curves,
Student's t tests for WT versus Ikk+/![]() mice. [View
Larger Version of this Image (43K GIF file)]
mice. [View
Larger Version of this Image (43K GIF file)]
To further assess the effect of a
reduction in Ikk![]() gene dose, mice were fed a diet high in fat. Fasting glucose and insulin
concentrations were consistently lower in Ikk
gene dose, mice were fed a diet high in fat. Fasting glucose and insulin
concentrations were consistently lower in Ikk![]() +/
+/![]() compared to Ikk
compared to Ikk![]() +/+
littermates (Fig.
5, E and F). Reduced insulin concentrations in the Ikk
+/+
littermates (Fig.
5, E and F). Reduced insulin concentrations in the Ikk![]() +/
+/![]() mice were maintained throughout glucose tolerance testing in 18-week-old
littermates (Fig.
5H). Dietary intake and weights were indistinguishable between
all pairs of Ikk
mice were maintained throughout glucose tolerance testing in 18-week-old
littermates (Fig.
5H). Dietary intake and weights were indistinguishable between
all pairs of Ikk![]() +/
+/![]() and Ikk
and Ikk![]() +/+
littermates. These data demonstrate that a reduction in Ikk
+/+
littermates. These data demonstrate that a reduction in Ikk![]() gene dose reduces fasting glucose and insulin concentrations and protects
against the development of insulin resistance in predisposed rodents.
gene dose reduces fasting glucose and insulin concentrations and protects
against the development of insulin resistance in predisposed rodents.
Our findings demonstrate that increased
IKK activity promotes insulin resistance, in obese rodents (12) when
the kinase is overexpressed, or when IKK is activated by known
stimulators. Conversely, reductions either in IKK activity or in the
expression of its IKK![]() subunit significantly improved insulin sensitivity. Even a 50%
reduction in gene dosage improved in vivo glucose and lipid
metabolism, which may explain why weak inhibitors of IKK
subunit significantly improved insulin sensitivity. Even a 50%
reduction in gene dosage improved in vivo glucose and lipid
metabolism, which may explain why weak inhibitors of IKK![]() ,
such as aspirin and sodium salicylate, have significant effects on
glucose and lipid homeostasis. Although not recognized previously, there
is an overlap between stimuli that activate IKK and conditions that
promote insulin resistance, including proinflammatory cytokines such
as TNF-
,
such as aspirin and sodium salicylate, have significant effects on
glucose and lipid homeostasis. Although not recognized previously, there
is an overlap between stimuli that activate IKK and conditions that
promote insulin resistance, including proinflammatory cytokines such
as TNF-![]() ,
hyperglycemia, phorbol esters and protein kinase C (PKC) enzymes,
Ser-Thr phosphatase inhibitors, and bacterial lipopolysaccharide.
These are either in vivo mediators of insulin resistance or
experimental mimics in cultured cells. Our findings are consistent
with potential links between chronic subacute inflammation and
insulin resistance (26, 27),
whether this is mediated by TNF-
,
hyperglycemia, phorbol esters and protein kinase C (PKC) enzymes,
Ser-Thr phosphatase inhibitors, and bacterial lipopolysaccharide.
These are either in vivo mediators of insulin resistance or
experimental mimics in cultured cells. Our findings are consistent
with potential links between chronic subacute inflammation and
insulin resistance (26, 27),
whether this is mediated by TNF-![]() produced in fat (28-31)
or through TNF-
produced in fat (28-31)
or through TNF-![]() -independent
mechanisms. As a potentially important example of the latter, in
rodent muscle, FFA infusion activates PKC-
-independent
mechanisms. As a potentially important example of the latter, in
rodent muscle, FFA infusion activates PKC-![]() (32), a
known activator of IKK (33), and
FFA-induced insulin resistance is suppressed by aspirin treatment and
in Ikk
(32), a
known activator of IKK (33), and
FFA-induced insulin resistance is suppressed by aspirin treatment and
in Ikk![]() +/
+/![]() mice (34).
IKK activation through any mechanism initiates NF-
mice (34).
IKK activation through any mechanism initiates NF-![]() B-mediated
transcription, which in certain cells would enhance the production
of TNF-
B-mediated
transcription, which in certain cells would enhance the production
of TNF-![]() .
This positive feedback loop could perpetuate a vicious cycle of
low-level inflammatory signaling, leading to insulin resistance. Our
findings predict that IKK inhibition breaks this cycle. Too few
tools are currently available to treat patients with insulin
resistance and type 2 diabetes; IKK
.
This positive feedback loop could perpetuate a vicious cycle of
low-level inflammatory signaling, leading to insulin resistance. Our
findings predict that IKK inhibition breaks this cycle. Too few
tools are currently available to treat patients with insulin
resistance and type 2 diabetes; IKK![]() may provide a valuable target for the discovery of new drugs to
treat these conditions.
may provide a valuable target for the discovery of new drugs to
treat these conditions.
REFERENCES AND NOTES
|
1. |
E. Kopp and S. Ghosh, Science 265, 956
(1994) [ISI][Medline]
. |
|
2. |
M. J. Yin, Y. Yamamoto, R. B. Gaynor, Nature 396,
77 (1998) [CrossRef][ISI][Medline]
. |
|
3. |
W. Ebstein, Berl. Klin. Wochnschr. 13, 337
(1876) . |
|
4. |
R. T. Williamson, Br. Med. J. 1, 760 (1901)
. |
|
5. |
J. Reid, A. I. Macdougall, M. M. Andrews, Br. Med. J.
2, 1071 (1957) . |
|
6. |
S. G. Gilgore, Diabetes 9, 392 (1960) [ISI]
. |
|
7. |
S. H. Baron, Diabetes Care 5, 64 (1982) [Abstract]
. |
|
8. |
Harvested liver and plantaris muscle samples were powdered
under liquid nitrogen and homogenized in ice-cold buffer [30 mM Hepes (pH
7.4), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM
phenylmethylsulfonyl fluoride (PMSF), 3 µM aprotinin, 10 µM
leupeptin, 5 µM pepstatin A, 25 mM benzamidine, 2 mM sodium
orthovanadate, 5 mM |
|
9. |
J. F. Caro, L. G. Dohm, W. J. Pories, M. K. Sinha, Diabetes
Metab. Rev. 5, 665 (1989) [ISI][Medline]
. |
|
10. |
L. J. Goodyear, et al., J. Clin. Invest. 95,
2195 (1995) [ISI][Medline]
. |
|
11. |
Supplementary Web material is available on Science
Online at www.sciencemag.org/cgi/content/full/293/5535/1673/DC1. |
|
12. |
J. Lee, M. Yuan, L. Hansen,
S. E. Shoelson, unpublished data. |
|
13. |
Murine 3T3-L1 fibroblasts (American Type Culture
Collection) were maintained in Dulbecco's modified Eagle's medium (DMEM)
containing 25 mM glucose and 10% calf serum under 5% CO2 in
air. Confluent monolayers were differentiated into adipocytes by treatment
for 3 days with DMEM containing 10% fetal bovine serum (FBS), insulin
(10 µg/ml), 1.0 mM dexamethasone, and 0.5 mM
isobutyl-1-methylxanthine under 10% CO2. Cells were returned to
DMEM containing 10% FBS and used for experiments 8 to 13 days after
the initiation of differentiation. Before these experiments, differentiated
3T3-L1 adipocytes were serum-starved for 16 hours in DMEM containing
0.1% bovine serum albumin (BSA). Stocks (1.0 M) of acetylsalicylic acid
(ASA) in 1.0 M tris-HCL were freshly prepared and pH was corrected to
7.4. Adipocytes were pretreated with 5 or 10 mM ASA for
2 hours at 37°C and then treated with either 6 nM murine TNF- |
|
14. |
M. Karin and M. Delhase, Semin. Immunol. 12,
85 (2000) [CrossRef][ISI][Medline]
. |
|
15. |
J. A. DiDonato, M. Hayakawa, D. M. Rothwarf, E. Zandi, M.
Karin, Nature 388, 548 (1997) [CrossRef][ISI][Medline]
. |
|
16. |
E. W. Harhaj and S. C. Sun, J. Biol. Chem. 272,
5409 (1997) [Abstract/Free Full Text] . |
|
17. |
K. A. Robinson, K. P. Boggs, M. G. Buse, Am.
J. Physiol. 265, E36 (1993) [Abstract/Free Full Text] . |
|
18. |
K. Paz, et al., J. Biol. Chem. 272,
29911 (1997) [Abstract/Free Full Text] . |
|
19. |
N. Konstantopoulos, S. E. Shoelson, unpublished
data. |
|
20. |
Fao hepatoma cells, grown to 80% confluency, were
serum-starved for 16 hours at 37°C in RPMI media containing 0.1% BSA.
Stock solutions of 1.0 M aspirin and 1.0 M sodium salicylate were freshly
prepared in 1.0 M tris-HCl (pH 7.4); stock solutions of 10 mM
ibuprofen, 10 mM naproxen, and 10 mM NS-398 (Biomol, Plymouth, PA)
were freshly prepared in ethanol. |
|
21. |
|
|
22. |
Cox1 |
|
23. |
E. Zandi, D. M. Rothwarf, M. Delhase, M. Hayakawa, M.
Karin, Cell 91, 243 (1997) [ISI][Medline]
. |
|
24. |
Q. Li, D. Van Antwerp, F. Mercurio, K. F. Lee, I. M.
Verma, Science 284, 321 (1999) [Abstract/Free Full Text] . |
|
25. |
Z. W. Li, et al., J. Exp. Med. 189,
1839 (1999) [Abstract/Free Full Text] . |
|
26. |
J. C. Pickup and M. A. Crook, Diabetologia 41,
1241 (1998) [CrossRef][ISI][Medline]
. |
|
27. |
A. Festa, et al., Circulation 102, 42
(2000) [Abstract/Free Full Text] . |
|
28. |
C. H. Lang, C. Dobrescu, G. J. Bagby, Endocrinology
130, 43 (1992) [Abstract]
. |
|
29. |
G. S. Hotamisligil, N. S. Shargill, B. M. Spiegelman, Science
259, 87 (1993) [ISI][Medline]
. |
|
30. |
R. Feinstein, H. Kanety, M. Z. Papa, B. Lunenfeld, A.
Karasik, J. Biol. Chem. 268, 26055 (1993) [Abstract/Free Full Text] . |
|
31. |
G. S. Hotamisligil, A. Budavari, D. Murray, B. M. Spiegelman,
J. Clin. Invest. 94, 1543 (1994) [ISI][Medline]
. |
|
32. |
M. E. Griffin, et al., Diabetes 48,
1270 (1999) [Abstract]
. |
|
33. |
Z. Sun, et al., Nature 404, 402
(2000) [CrossRef][ISI][Medline]
. |
|
34. |
J. K. Kim, et al., J. Clin. Invest. 108,
437 (2001) [Abstract/Free Full Text] . |
|
35. |
This manuscript is dedicated to the memory of Shirley
Blauner. We thank T. Maratos-Flier for helpful discussions and
J. Warram for assistance with statistical analyses. Supported by NIH
grants DK51729 (S.E.S.), DK45493 (S.E.S.), and AI43477 (M.K.); a research
grant (S.E.S.) and a Mentor-Based Fellowship (M.Y. and N.K.) from the
American Diabetes Association; the Cancer Research Institute (Z.W.L.); an
American Cancer Society Research Professorship (M.K.); and a Burroughs
Wellcome Fund Scholar Award in Experimental Therapeutics (S.E.S.). |
12 April 2001; accepted 12 June 2001
10.1126/science.1061620
Include this information when citing this paper.